
RAS: un sistema pieno di sorprese
Enrico Bologna
Specialista in Medicina Interna, Gastroenterologia e Patologia generale.
Già Primario Ospedale Fatebenefratelli, Isola Tiberina, Roma.
Libero docente in Patologia Medica, Università di Roma “Sapienza”
ABSTRACT
RAS: an ever amazing system.
In the last decade the results of many studies indicate that the primary effector hormone of RAS, angiotensin II, not only mediates unfavorable effects on cardiovascular system, but also is implicated in physiological and pathological functions of many biological systems. Some angiotensin II-derived peptides, as angiotensin(1-7), angiotensin(1-9) and others, activate several receptors promoting signaling cascades that contribute both to normal functions and disease progression. This short review focused on these new acquirements and the possible therapeutic venues.
RIASSUNTO
Nell’ultimo decennio i risultati di numerose ricerche hanno permesso di modificare profondamente il classico concetto di RAS come sistema basato sulla formazione di Angiotensina II e in quanto tale potenziale responsabile di effetti sfavorevoli sull’apparato cardiovascolare. Il RAS viene oggi considerato come un sistema che agisce su molteplici apparati mediante due vie principali, attivate rispettivamente da ACE e ACE2, con formazione di numerosi peptidi capaci di agire su vari recettori. Da alcuni di questi peptide, in particolare angiotensina(1-7) e angiotensina(1-9), e da nuovi agonisti dei diversi recettori si possono attendere importanti progressi in terapia.
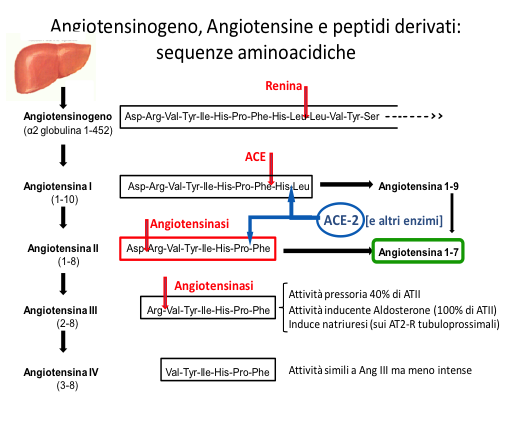
In una visione classica il Sistema Renina-Angiotensina (Renin Angiotensin System, RAS) è costituito dalla Prorenina, prodotta da vari tessuti (surrene, ovaia, testicolo, placenta e retina), che nelle cellule renali iuxtaglomerulari subisce una scissione proteolitica con rimozione di 43 aminoacidi dando luogo all’enzima aspartil-proteasi Renina; questa scinde una alfa-2 globulina costituita da 452 aminoacidi prodotta dal fegato, l’Angiotensinogeno, a formare un decapeptide inattivo denominato Angiotensina I (Ang I) che è successivamente scisso da un enzima di conversione (Angiotensin Converting Enzyme, ACE) a formare l’octapeptide attivo Angiotensina II (Ang II). Gli effetti più noti di Ang II dipendono dal suo legame al recettore AT1 (AT1R). Questo appartiene alla famiglia dei recettori transmembrana accoppiati a proteine G (G Protein Coupled Receptor, GPCR), che rispondono al contatto con molecole extracellulari attivando la trasduzione di segnali che determinano risposte cellulari. Il legame di Ang II ad AT1-R determina l’emissione di molteplici segnali che provocano l’attivazione del fattore nucleare NFkb, mediatore ubiquitario di risposte di tipo infiammatorio comprendenti fra l’altro la liberazione di proteine di adesione (VCAM-1, ICAM-1, MCP-1, E-selectin); altre conseguenze di questo legame sono la produzione di specie reattive dell’ossigeno, l’ipertrofia dei miociti vascolari con costrizione arteriosa, l’aumento della pressione arteriosa e l’induzione di insulinoresistenza. Dall’attivazione di AT1-R dipende anche l’espressione di recettori delle LDL ossidate (Lecitin-like OX LDL receptor, LOX) e la liberazione di citochine e della Matrix MetalloProteinase (MMP), ritenuta la principale responsabile dell’instabilità delle placche ateromasiche23.
Sul rene il legame di Ang II ad AT1-R provoca attivazione dei canali del calcio voltaggio-dipendenti con contrazione arteriolare generalizzata, che è particolarmente intensa a carico delle arteriole efferenti; determina inoltre riduzione del flusso midollare. Queste alterazioni modificano l’equilibrio delle forze di Starling determinando aumento del riassorbimento idrosalino21.
Nel sistema nervoso centrale Il RAS esercita, in modo indipendente dal RAS sistemico, un ruolo di grande rilievo sulle funzioni cardiovascolari influenzando il sistema nervoso autonomo, la sensibilità barorecettoriale, la liberazione di vasopressina, la sete e la ricerca del sale. Ang II agisce anche come neuropeptide aumentando, attraverso il legame agli AT1-R, l’eccitabilità dei neuroni dei centri regolatori dell’ipotalamo e del mesencefalo17. Inoltre, le alterazioni circolatorie indotte da Ang II e la produzione di specie reattive dell’ossigeno determinano una serie di effetti sfavorevoli fra cui disfunzione endoteliale con riduzione delle cellule progenitrici endoteliali, riduzione della vasodilatazione flusso-mediata, alterazione del coupling neurovascolare, aumento della permeabilità della barriera emato-encefalica, infiammazione e ipertrofia vascolare. Queste alterazioni determinano ischemia cerebrale e predispongono all’ictus e alla demenza5,25.
L’attivazione dei recettori AT1 ad opera di Ang II è condizionata da variabili genetiche, fisiologiche e farmacologiche. Per quanto riguarda le prime, il gene umano di ACE, posto sul cromosoma 17, è soggetto a polimorfismi per Inserimento (allele-I) o Delezione (allele-D), quindi con possibilità di tre genotipi: DD, ID, II.
La concentrazione plasmatica e tessutale (in particolare cardiaca) di ACE, e la conseguente produzione di Ang II, è 1,5 – 3 volte più elevata nei soggetti con il genotipo DD rispetto a quelli con genotipo II; aumenti di entità intermedia si rilevano per il genotipo ID. Questi rilievi concordano con l’osservazione che, in soggetti normotesi, l’affetto acuto di un ACE-inibitore è significativamente maggiore e più protratto in presenza di ACE genotipo II.
Numerose ricerche hanno dimostrato che la presenza di genotipo ACE DD si associa a dilatazione e rimodellamento ventricolari postinfartuali, ipertrofia ventricolare sinistra in soggetti ipertesi, ipertrofia ventricolare sinistra in soggetti con cardiomiopatia ipertrofica, ipertrofia ventricolare sinistra da allenamento sportivo, ridotta sopravvivenza e maggior massa ventricolare in soggetti con insufficienza cardiaca da cardiomiopatia dilatativa idiopatica, maggior rischio di cardiomiopatia negli etilisti, maggior rischio aterosclerotico in soggetti diabetici e dislipidemici. Infine, l’espressione dei recettori AT1 è influenzata da vari fattori endogeni ed esogeni: è favorita da LDL, Insulina, Progesterone, ed Eritropoietina mentre è ostacolata da Angiotensina II, Interferone γ, Estrogeni, Vitamina A, EGF, PDGF, Ormone tiroideo, NO e Statine7,22,33.
Agli effetti dell’attivazione di AT1-R si contrappongono quelli conseguenti all’attivazione che Ang II svolge nei confronti dei recettori AT2, anch’essi appartenenti ai GCPR e costituiti da una sequenza aminoacidica che ripete per il 34% quella degli AT1-R umani e per circa l’80% quella degli AT1-R dei roditori, aspetto questo che conferisce particolare valore agli studi sperimentali condotti su questi animali.
Il legame di Ang II agli AT2-R induce vasodilatazione (via BK, NO, cGMP), stimola fosfatasi inibitorie di ATR-1, contrasta il signaling infiammatorio di AT1-R, inibisce la crescita cellulare, ostacola il rimodellamento vascolare e interviene favorevolmente nella differenziazione neuronale10,13. E’ interessante rilevare che i recettori AT2 presentano una concentrazione particolarmente elevata nel sistema nervoso centrale, il cui sistema RAS è indipendente da quello periferico e in cui i livelli di Ang II sono più elevati di quelli rilevati nel sangue circolante. Questi recettori sono presenti in aree preposte al controllo del bilancio idro-elettrolitico, della secrezione di Vasopressina, del sistema autonomo e delle attività cognitive, comportamentali e motorie. In particolare, i recettori AT2 intervengono in varie funzioni cerebrali svolgendo attività protettiva soprattutto in rapporto a malattie neurodegenerative (Alzheimer, Parkinson), epilessia, diabete mellito e sindrome metabolica. L’attività protettiva dei recettori AT2 ha trovato conferma sperimentale nella capacità di un agonista (“Composto 21”) di ridurre i danni vascolari e la fibrosi in ratti spontaneamente ipertesi29.
Il blocco farmacologico di RAS può essere effettuato a più livelli con molecole che inibiscono la Renina (Aliskiren), l’ACE (ACE-inibitori) o il recettore AT1 (Angiotensin Receptor Blocker, ARB). Ma la somministrazione associata di un ACE inibitore (Ramipril) e dì un ARB (Telmisartan) non ha determinato vantaggi in soggetti vasculopatici o diabetici con danno d’organo, come dimostrato dallo Studio ONTARGET. Inoltre itentativi di associare un inibitore diretto della renina (Aliskiren) con un ACE inibitore o con un ARB sono risultati in un peggioramento della prognosi di pazienti diabetici, come osservato nello Studio ALTITUDE. Sulla base di un’ampia meta-analisi12l’EMA (European Medicines Agency) ha avviato nel 2013 una rivalutazione della pratica di associare medicinali che bloccano a diversi livelli il RAS nel trattamento della ipertensione e della insufficienza cardiaca congestizia.Per quanto riguarda il blocco isolato della renina, nello Studio ALPINE, presentato al meeting 2013della American Society of Hypertension, il trattamento per 36 settimane con Aliskiren di pazienti con ateromasia aortica è stato seguito da un significativo aumento volumetrico delle placche aortiche rispetto ai controlli, mentre nessuna variazione è stata rilevata in pazienti trattati con ACEi o ARB24. Questo sorprendente risultato contrasta con le dimostrazioni di effetto protettivo di Aliskiren nei confronti del processo aterosclerotico. Si deve però considerate che queste ultime dimostrazioni, tutte derivanti da studi sperimentali su animali, sono state effettuate su fasi precoci dell’aterosclerosi sperimentalmente indotta e quindi in condizioni completamente differenti rispetto allo studio ALPINE, che è stato condotto su lesioni aterosclerotiche umane in stato avanzato.
Un altro aspetto di grande interesse emerso dallo studio ALPINE è rappresentato dal netto aumento della Leptina osservato nei pazienti trattati con Aliskiren. Molti studi hanno dimostrato che Leptina è un predittore indipendente di eventi acuti coronarici e cerebrali36,37; è stato anche osservato che i livelli di Leptina si riducono a seguito di trattamento con ACE-inibitori o ARB, mentre non si modificano in misura significativa per effetto di calcio-antagonisti o di statine17.
Questi risultati, insieme a nuove acquisizioni provenienti dalle ricerche di base, dimostrano la grande complessità degli effetti dei vari componenti del RAS in molti tessuti, in buona parte spiegabili con le potenzialità pleiotropiche tipiche dei GPCR. Tale complessità ha trovato una possibile spiegazione nella scoperta di prodotti di degradazione di Ang II come Ang 2-8 (Ang III) e Ang 3-8 (Ang IV) e nella successiva identificazione di altri peptidi biologicamente attivi derivati da Ang II come Ang 1-9 e Ang 1-7 così come di altri recettori oltre ad AT2-R come AT3-R, AT4-R (o Insulin Regulated Aminopeptidase, IRAP) e Mas. Sono stati inoltre identificati numerosi altri enzimi responsabili della scissione di peptidi quali catepsine, callicreina, pepsina, TPA, chimasi e le aminopeptidasi A e N, da cui dipende la produzione di Ang III e Ang IV; maggiore interesse riveste la carbossipeptidasi ACE2, da cui dipende la produzione di Ang 1-7 18.
In conseguenza di queste acquisizioni il concetto secondo cui Ang II è il principale peptide effettore del RAS e che il suo ruolo primario è rappresentato dall’attività regolatoria sul tono arterioso è attualmente da ritenersi superato; il RAS deve invece essere considerato come un sistema altamente complesso che influenza con molteplici meccanismi e con effetti spesso contrastanti non solo il sistema cardiovascolare, i reni e i surreni, ma anche altre strutture fra cui il sistema nervoso e il midollo emopoietico.
Tra i peptidi derivanti dalla scissione di Ang II particolare interesse è stato sollevato dall’eptapeptide Ang (1-7), prodotto dall’azione di ACE2 su Ang I (via Angiotensina 1-9) e da Ang II. Ang (1-7), inizialmente considerato come un derivato inattivo di Ang II, si è rivelato come un importante componente del RAS; esso infatti agisce sul recettore Mas, la cui attivazione svolge effetti opposti a quelli determinati dal recettore AT16,30,31: esso infatti provoca vasodilatazione liberando NO e prostaciclina e contrasta le attività mitogene, procoagulanti, aritmogene, sodioritentive e antidiuretiche di Ang II. In ratti con deficit geneticamente indotto del recettore Mas si osservano importanti modificazioni fenotipiche, quali disfunzione endoteliale, alterazioni della pressione arteriosa e dei baroriflessi, fibrosi cardiaca, trombofilia e alterazioni simil-sindrome metabolica. Nell’animale la presenza del recettore Mas è particolarmente rilevante nel sistema nervoso centrale, in corrispondenza di aree coinvolte nel controllo di funzioni cardiovascolari oltre che di funzioni comportamentali3. Si può fra l’altro ritenere che una parte degli effetti favorevoli svolti dagli ACE-inibitori dipenda dalla maggior produzione di Ang (1-7) resa possible dal maggior accumulo di Angiotensinogeno.
Indipendentemente dall’intervento del recettore Mas, inoltre, Ang (1-7) potenzia l’attività di bradichinina e antagonizza l’effetto ipertrofizzante di Ang II esercitando una ampia azione inibente l’adesione di cellule all’endotelio e quindi una potente attività antiaterogena e vasoprotettrice4,18,34.
Alla identificazione delle molteplici azioni protettive svolte da Ang (1-7) si è aggiunta la dimostrazione sperimentale della capacità di AVE 0991, agonista non peptidico che mima gli effetti di Ang (1-7), di prevenire l’aterosclerosi sperimentale35. In studi successivi è stato dimostrato che AVE 0991 inibisce l’espressione di NADPH indotta dal legame di Ang II al recettore AT1, con conseguente blocco della produzione di ROS e mantenimento della produzione di NO; AVE 0991 inoltre riduce l’espressione di molecole infiammatorie da parte di macrofagi e di T-linfociti con conseguente riduzione della concentrazione plasmatica di IL-6, IL-12, SAA e MCP-115,16. Di questo agonista è in corso di valutazione l’impiego su stent9.
Gli effetti benefici dell’attivazione del recettore Mas hanno trovato conferma in uno studio condotto su un altroagonista di questo recettore, CGEN-856S, che si è dimostrato capace di contrastare efficacemente l’infarto miocardiaco sperimentale e il rimodellamento cardiaco32.
Considerando l’esistenza di due tipi di ACE e di due mediatori derivati dall’angiotensinogeno, Ang II e Ang (1-7), il RAS viene attualmente concepito come un sistema costituito da due vie contrapposte: ACE – Ang II –AT1R e ACE2 – Ang (1-7) – Mas (Tab. 2). Ang (1-7) viene prodotta ad opera di ACE2 sia da Ang I sia, molto più efficacemente, da Ang II. Durante trattamento con ARBs una quota maggiore di Ang II può essere resa disponibile per attivare i recettori AT2 o per essere convertita in Ang (1-7)14. I farmaci ACEi determinano un aumento della concentrazione plasmatica di Ang (1-7) di circa 10 volte, e questo potrebbe essere un meccanismo che contribuisce agli effetti benefici di questi farmaci. D’altra parte l’espressione di mRNA codificante per ACE2 aumenta di 5 volte in corso di trattamento con ACEi e di 3 volte durante trattamento con ARB8.
L’asse Ang II – Ang (1-7) – Mas è attualmente oggetto di ricerca per l’individuazione e la sperimentazione di molecole in grado di stimolarne l’attivazione. rappresentando così un nuovo mezzo per ostacolare l’aterogenesi.
Ang III è attualmente considerata come la principale attivatrice di AT2R nel tubulo prossimale, dove media l’effetto natriuretico 28.
Ed è recentissima l’identificazione, nel sangue umano e di roditori, di un eptapeptide che differisce dalla Angiotensina (1-7) per l’aminoacido in posizione 1 (alanina in luogo di acido aspartico) e che esercita azioni analoghe a quelle di Angiotensina (1-7). Questo peptide sarebbe prodotto nel miocardio e la sua attività non appare mediata del recettore Mas20. Un ultimo aspetto che merita di essere segnalato riguarda l’esistenza e le potenzialità dei recettori della prorenina, identificati circa 10 anni fa nelle cellule mesangiali umane. Tali recettori, denominati (pro)renin receptor, (P)RR in quanto capaci di legarsi tanto a Prorenina quanto a Renina, sono presenti sia alla superficie cellulare sia nei compartimenti intracellulari, soprattutto perinucleari38. L’affinità di Prorenina per i PRR è 3-4 volte maggiore di quella di Renina1,27; è pertanto verosimile che Prorenina possa agire soprattutto nei tessuti in sede sia intra- sia extracellulare, dove può raggiungere concentrazioni sufficientemente elevate da attivare segnali dipendenti e indipendenti da Ang II2. All’attività dei (P)RR può in ipotesi essere attribuita la responsabilità dei già ricordati effetti sfavorevoli inattesi in risposta alla somministrazione di Aliskiren, che inibendo la formazione di Renina può determinare accumulo di Prorenina e conseguente attivazione dei PRR38. In accordo con questa ipotesi sta la recente osservazione che la sovraespresione sperimentalmente indotta del (P)RR determina nel ratto fibrosi e disfunzione cardiaca, effetto questo che non appare mediato dall’intervento di Ang II, poiché non viene ridotto dal blocco farmacologico degli AT1-R26.
Tab. 1
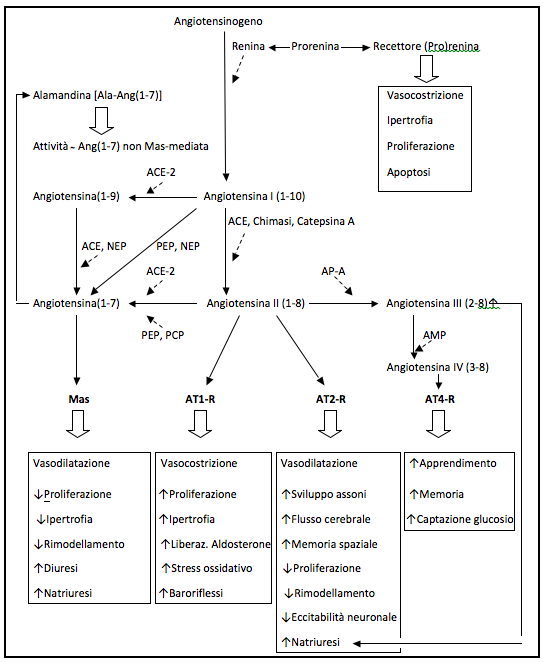
Tab. 2 (Modif da 11) ACE: Angiotensin Converting Enzyme; PEP: Phospho Enol Pyruvate; Nep: Neutral EndoPeptidase; AP-A: Aminopeptidase-A; AMP: Adenosine Mono Phosphate; PCP: PolyCarboxyl Peptidase.
BIBLIOGRAFIA
1 Batenburg WW, Krop M, Garrelds IM & al: Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens. 2007:25,2441
2 Batenburg WW, Lu X, Leijten F & al: Renin- and prorenin-induced effects in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor: does (pro)renin-(pro)renin receptor interaction actually occur? Hypertens. 2011:58,1111]
3 Becker LK, Etelvino GM, Walther T& al: Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am J Physiol Heart Circ Physiol. 2007:293;H1416
4 Benter IF, Yousif MH, Cojocel C & al: Angiotensin(1-7) prevents diabetes-induced carsdiovascular dysfunction. Am J Physiol Heart Circ Physiol 2007:292,H666
5 De Silva TM, Faraci FM: Effects of angiotensin II on the cerebral circulation: role of oxidative stress. Front Physiol 2013:3,484
6 Douglas M, Hsieh F, Baronas F & al: A novel angiotensin converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000:87,E1
7 Fernández-Solà J, Nicolás JM, Oriola J& al: Angiotensin-converting enzyme gene polymorphism is associated with vulnerability to alcoholic cardiomyopathy. Ann Intern Med. 2002:137,321.
8Ferrario CM, Jessup J, Chappell MC & al: Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin converting enzyme 2. J Circulation 2005:111,2605.
9 Francione V, Befani A, Savoia C & al: Ilk sistema ACE2-Ang(1-7)-recettore Mas: una nuova frontiera nella fisiopatologia del rimodellamento cardiovascolare. Ipertens Prevens Cardiovasc 2012:19,217
10 Gallo-Payet N, Schum M, Baillargeon JP & al: AT2 receptor agonists exploiting the beneficial armo f Ang II signaling. Curr Iperten rew 2012:8,47
11 Guimond M-O. Gallo-Payet N: The angiotensin II type receptor in brain functions. An update. Internat J Hypertens epun 2012, dec 25
12 Harel Z, Gilbert C, Wald R & al: The effect of combination treatment with aliskiren and blockers of the renin-angiotensin system on hyperkaliemia and acute kidney injury: systematic review and meta-analysis- BMJ 2012:344,e42
13 Horiuchi M, Jwanami J, Mogi M: Regulation of angiotensin II receptors beyond the classical pathway. Clin Sci 2012:123,193.
14 Iwai M, Horiuchi M: Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs ACE2-angiotensin(1-7)-Mas receptor axis. Hypertens Res 2009:32,533
15 Jawien J, Toton-Zuranska J, Gajda M& al: Angiotensin-(1-7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J Physiol Pharmacol. 2012:63,77.
16 Jawien J, Toton-Zuranska J, Kus K, The effect of AVE 0991, nebivolol and doxycycline on inflammatory mediators in an apoE-knockout mouse model of atherosclerosis. Med Sci Monit. 2012:Oct;18, BR389.
17 Koh KK, Park SM, Quon MJ & al: Leptin and cardiovascular disease: response to therapeutic intervention. Circulation 2008:117,3238
18 Kuba K, Imai Y, Penninger JM: Multiple function of angiotensin converting enzyme 2 and its relevance in cardiovascular disease. Circ J 2013:77,301
19 Kucharewicz I, Pawlak R, Matys T 6 al: Antithrombotic effect of captopril and losartan is mediated by angiotensin (1-7). Hypertension 2002:40,774
20 Lautner RQ, Villela DC, Fraga-Silva RA& al: Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013:112,1104
21 Lees KR, MacFaiden RJ, Doig JK & al: Role of angiotensin in the extravascular system. J Hum Hypertens. 1993 Aug 7,Suppl 2:S7.
22 McNamara DM, Holubkov R, Janosko K& al: Pharmacogenetic interactions between beta-blocker therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. Circulation. 2001:103,1644.
23 Metha PK, Griendling KK: Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007:292,C82
24 Mihai G, Varghese J, Kampfrath T& al: Aliskiren effect on plaque progression in established atherosclerosis using high resolution 3D MRI (ALPINE): A Double-Blind Placebo-Controlled Trial. J Am Heart Assoc. 2013 May 17;2(3):e004879
25 Mogi M, Iwanami J, Horiuchi M: Roles of brain angiotensin II in cognitive function and dementia. Internat J Hypertens epub 2012, dec 11
26 Moilanen AM, Rysä J, Serpi R & al: (Pro)renin receptor triggers distinct angiotensin II-independent extracellular matrix remodeling and deterioration of cardiac function. PLoS One 2012: 7,e41404
27 Nabi AHMN, Kageshima A, Uddin M & al: Binding properties of rat prorenin and renin to the recombinant rat renin/prorenin receptor prepared by a baculovirus expression system. Internat J Mol Medicine. 2006:18,483
28 Raizada MK, Ferreira AJ: ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol 2007:50,112
29 Rehman A, Leibowitz A, Yamamoto N& al: Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 2012 F59,291
30 Santos RA, Simoes e Silva AC, Maric C & al: Angiotensin(1-7) is an endogenous ligand rof the G protein-coupled receptor MAS. Proc Natl Acad Sci USA 2003:100,8258
31 Santos RA, Ferreira AJ, Braga TV & al: Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol 2013:216,R1
32 Savergnini SQ, Ianzer D, Carvahlo BL & al: The novel Mas agonist, CGEN-856S, attenuates isoproterenol-induced cardiac remodeling and myocardial infarction injury in rats. Plos One 2013:8,e57757
33 Seckin D, Ilhan N, Ilhan N& al: The relationship between ACE insertion/deletion polymorphism and coronary artery disease with or without myocardial infarction. Clin Biochem. 2006:39,50
34 Tesanovic S, Vinh A, Gaspari TA & al: Vasoprotective and atheroprotective effects of angiotensin(1-7). Arterioscler Thromb Vasc Biol 2010:30,1606
35 Toton-Zuranska J, Gajda M, Pyka-Fosciak G& al: AVE 0991-angiotensin-(1-7) receptor agonist, inhibits atherogenesis in apoE-knockout mice. J Physiol Pharmacol. 2010:61,181.
36 Wallace AM, McMahon AD, Packard CI & al: Plasma leptin and the rrisk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 2001:104,3052
37 Wallerstedt SM, Eriksson AL, Niklason A & al: Serum leptin and myocardial infarction in hypertension. Blood Press 2004:13,243
39 Wencheng Li, Hua P, Dale M & al: The Prorenin and (Pro)renin Receptor: new players in the brain renin-angiotensin system? Internat J Hypertens 2012, epub dec 18.