

NOTIZIARIO Marzo 2011 N°3
"LE EVIDENZE SULLA SICUREZZA DEI FARMACI"
A cura di Giuseppe Di Lascio
Con la collaborazione di:
Bagalino Alessia, Bauzulli Doriana, Di Lascio Alessandro, Di Lascio Susanna, Levi Della Vida Andrea, Melilli Simonetta, Pallotta Pasqualino, Sesana Giovanna, Stazzi Claudio, Zimmatore Elena
LUTTO DEL MONDO SCIENTIFICO
Si è spento all’età di 79 anni Mario Condorelli

Venerdì 04 marzo 2011 è deceduto a Napoli a 79 anni Mario Condorelli, cardiologo di fama mondiale. Nato a Napoli da Luigi, insigne medico e scienziato di origine siciliana, si è laureato a Roma. È stato, di poi, al seguito di Giuseppe Zagari, clinico medico della facoltà di Medicina dell’Università partenopea, con un breve intervallo nell’Università di Messina. Napoli è stata anche la città d’origine della moglie Paola Chiariello, figlia di Alfonso Giovanni, fondatore della Clinica Mediterranea. Condorelli ha ricoperto diversi incarichi importanti istituzionali nell'Università degli Studi Federico II di Napoli ed anche politici. È stato senatore nella IX, X e XI legislatura, con l’impegno anche di sottosegretario alla Sanità nel governo Dini. È stato autore, editore di numerose opere scientifiche. Ha ricoperto anche la carica di Presidente del Consiglio superiore di sanità e di Presidente della Società italiana di medicina interna. La medaglia d'oro al merito della sanità pubblica e la nomina a Cavaliere di Gran Croce al merito della Repubblica Italiana sono tra i riconoscimenti più importanti ricevuti.
Alla famiglia vanno sentite condoglianze di tutta l’associazione AMEC.
Fatali gli errori con l’uso della morfina solubile orale
La FDA il 10 gennaio 2011 ha annunciato che, in occasione delle segnalazioni di eventi avversi gravi, sino al decesso, per overdose di morfina in rapporto alle prescrizioni in milligrammi (mg) per le soluzioni orali di 100 mg/5 ml (20 mg/mL), fraintese involontariamente come millilitri (ml), la Roxane Laboratories ha notificato agli operatori sanitari le variazioni sull'etichetta del prodotto. Quando questo tipo di errore si è verificato con la soluzione ad alta concentrazione, il risultato è stato un’overdose di 20 volte il voluto. Inoltre, altri errori di dosaggio sono derivati, in genere, per confusione o incomprensione della concentrazione di morfina in soluzione orale. Di fatto, la morfina solubile per via orale è ora disponibile in concentrazioni di 10 mg/5 ml, 20 mg/ml 5 e 100 mg/5 ml (20 mg/mL). Quelle di 100 mg/5 ml (20 mg/mL) sono indicate solo per il sollievo del dolore acuto e cronico, da moderato a grave, nei pazienti tolleranti gli oppioidi.
Sproporzione tra i decessi per overdose di metadone e il numero delle prescrizioni
Una rassegna nazionale ha dimostrato un numero sproporzionato di decessi da overdose accidentale di oppiacei, correlato all’uso di metadone.
Difatti, nel 30% di essi era coinvolto il metadone, mentre solo nel 5% un altro oppiaceo. Peraltro, i ricercatori, guidati da Webster Lynn di Salt Lake City, Utah, hanno osservato che i problemi sembravano centrati su quelle prescrizioni per il dolore e non per la disintossicazione da tossicodipendenza (American Academy of Pain Medicine (AAPM) 26th Annual Meeting: Poster 228. Presented February 3-5, 2010). Gli AA hanno anche annotato che idecessi, correlati al consumo di oppiacei, si erano elevati sino al 260% nel 2005 rispetto ai livelli registrati nel 2001. Di conseguenza, la FDA aveva già annunciato nel febbraio 2009 che sarebbero stati richiesti REMS per certi oppioidi per garantire che i loro benefici dovessero superare i rischi.Un gruppo di esperti, chiamato a studiare il problema, concludeva che uno dei motivi principali di danno ai malati era costituito dall’uso inappropriato delle tabelle di conversione utilizzate dai medici per il passaggio da un oppioide a un altro. Sarebbero state, così, raccomandate dosi eccessive di metadone per la maggior parte dei pazienti. Al riguardo c’è, poi, da considerare che altre condizioni, in genere, contribuiscono agli effetti lesivi del metadone, come le improvvise comorbidità mediche e psichiatriche, tra cui l’uso di sostanze e di altri farmaci agenti sul sistema nervoso centrale, come l’alcool, le benzodiazepine o gli antidepressivi.
Tigeciclina ad alto rischio di eventi avversi e di morte
La tigeciclina (tygacil, Pfizer) è un antibiotico di prima classe, ad ampio spettro, del gruppo delle glicilcicline che agisce bloccando la crescita dei batteri.È stata approvata dalla FDA (US Food and Drug Administration) per il trattamento delle infezioni complicate intra-addominali, quelle complicate della cute, la polmoniteacquisita in comunità, le infezioni da stafilococco aureo meticillino-resistente (MRSA) o enterococco vancomicino-resistente (VRE). Ha dimostrato anche efficacia nella polmonite ospedaliera associata alla ventilazione meccanica e nella batteriemia, nello shock settico e nelle infezioni del tratto urinario. È, peraltro, attiva contro gli agenti patogeni che le sono sensibili ma resistenti ad altri antibiotici. Rui Wang e collaboratori del PLA General Hospital, Beijing, People's Republic of Chinahanno condotto una meta-analisi sulla sicurezza della tigeciclina per chiarirne le proprietà in tema di efficacia e sicurezza (Antimicrob Agents Chemother. 2010 Dec 20). Gli AA., utilizzando i dati raccolti da otto studi randomizzati, controllati, di alta qualità hanno analizzato il successo clinico, cioè la risoluzione completa o il sostanziale miglioramento dei sintomi e dei segni d’infezione senza ulteriore terapia antimicrobica o intervento chirurgico per la malattia. Hanno anche verificato il successo microbiologico, cioè l'eradicazione degli agenti patogeni sulla base dei risultati clinici in cui le culture post-trattamento non erano state effettuate. Gli Autori hanno segnalato, quindi, che la tigeciclina in monoterapia era clinicamente efficace come la vancomicina e l’aztreonam per le infezioni complicate della cute e annessi, similarmente all’imipenem/cilastatina, al ceftriaxone più il metronidazolo per le infezioni complicate intra-addominali e come la levofloxacina per la polmonite acquisita in comunità. Tutto questo si è dimostrato valido sia nella popolazione clinicamente valutabile sia nell’intention-to-treat.Pur tuttavia, nonostante l'efficacia simile, la tigeciclina si è associata a un aumento dell'incidenza di tutti gli eventi avversi, come la febbre, il mal di testa, le infezioni, le addominalgie, i brividi e il dolore, e, in particolare, quelli che coinvolgono il sistema digestivo. Difatti, come negli studi precedenti, la nausea e la diarrea sono state gli eventi avversi più comuni. Iltassodieffetti avversicardiovascolari, invece, è risultato inferiore. Sulla base dell’insieme di tali dati, emerge, pertanto, la necessità per i medici di monitorare i segni e i sintomi, soprattutto digestivi, nei pazienti trattati con quest’antibiotico per il quale la meta-analisi ha anche mostrato numericamente tassi di mortalità, sia per tutte le cause sia possibilmente correlate al farmaco, più elevati nel confronto con gli altri antibiotici. Nel settembre 2010 l’US Food and Drug Administration ha, per l’appunto, diramato ai medici sul loro sito web un avviso di sicurezza sull’aumentato rischio di morte associato all'uso della tigeciclina, più evidente nei pazienti con polmonite acquisita in ospedale e in particolare con quella associata a ventilazione meccanica. La FDA ha, inoltre, citato i dati di un’analisi globale di 13 studi in cui 150 dei 3.788 pazienti, trattati con la tigeciclina, sonomorti, rispetto ai 110 dei 3.646 trattati con altri antibiotici (4,0% vs 3,0%). La differenza di rischio, aggiustato per tutte le cause di mortalità, in conformità a un modello a effetti casuali, è stata dello 0,6% tra la tigeciclina, rispetto ai controlli. In rapporto con quanto riportato, l'EMEA (Agenzia Europea dei Medicinali) ha ultimato il riesame del profilo rischio- beneficio del Tygacil nel corso della procedura di rinnovo dell'autorizzazione d'immissione in commercio, a cinque anni dalla prima. Il CHMP (Comitato per i Prodotti Medicinali per Uso Umano) dell'Agenzia ha concluso che i benefici del prodotto continuano a superare i rischi ma ha inteso raccomandare modifiche delle informazioni sull’antibiotico per il suo uso appropriato, indicando soprattutto l’aumentodella mortalità derivata dagli studi clinici. In particolare è stato redatto un profilo di raccomandazione per i medici prescrittori come segue:
• il tygacil è indicato solamente nelle infezioni complicate della cute e dei tessuti molli o nelle infezioni intra-addomminali complicate,
• il Tygacil va prescritto solo quando gli altri antibiotici non sono opportuni,
• il trattamento con Tygacil deve prevedere la stretta monitorazzazione, specialmente per accertare il possibile sviluppo di superinfezioni.
Da notare che l'insorgenza di superinfezioni, in particolare di polmonite, può associarsi a ridotta sopravvivenza del paziente e in tal caso bisogna passare a un altro antibiotico. Comunque, il prodotto è disponibile solo negli ospedali.
Complicazioni precoci della terapia con cediranib
Il cediranib, noto anche come AZD2171 e il cui nome commerciale provvisorio è recentin, è un potente inibitore orale tirosin-chinasico di VEGFR-1, VEGFR-2, VEGFR-3(vascular endothelial growth factor). È stato sviluppato dalla ditta Astra Zeneca come un possibile agente chemioterapico anti-cancro. Dal 2007 sono iniziati i test clinici in fase I per il trattamento del carcinoma del polmone non a piccole cellule, di quello del rene e colorettale negli adulti, così come dei tumori del sistema nervoso centrale nei bambini. Sono in corso anche studi collaterali delle interazioni con altri farmaci anticancro.
Giàil 27 febbraio 2008 l’AstraZeneca aveva, però, annunciato che l'uso del recentin nel carcinoma polmonare non a piccole cellule non sarebbe progredito alla fase III per aver fallito il suo obiettivo principale. L'8 marzo 2010 annunciava anche il fallimento del suo utilizzo in prima linea nel carcinoma colorettale metastatico, in ragione del confronto clinico con l’avastin(bevacizumab), leader del mercato in tale campo.
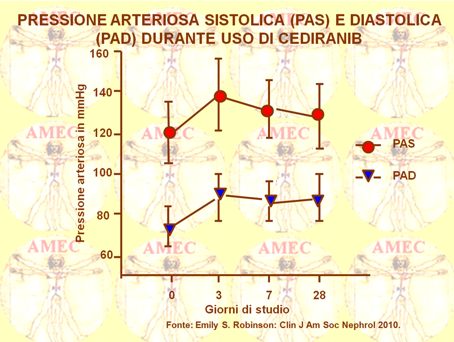
Emily S. Robinson e collaboratori del Brigham and Women's Hospital, Boston, considerando che il cediranib è più potente rispetto ai vecchi inibitori VEGF, ma teorizzando che potrebbe produrre effetti collaterali più marcati, hanno svolto uno studio tra il 2005 e il 2008 che ha coinvolto, per una media di 84 giorni, 46 donne di età tra i 41 e i 77 anni, con recidiva di carcinoma ovarico epiteliale (Clin J Am Soc Nephrol 2010). All’inizio cediranib è stato dosato a 45 mg, per poi passare a 30 per gli eccessivi effetti collaterali, in specie stanchezza e diarrea, dopo i primi 11 pazienti.
L’ipertensione si è sviluppata nel 67% dei casi entro i 3 giorni di cura, nel 73% entro i 7 e nell’87% a termine dello studio.Il 43 per cento ha sviluppato, peraltro, un’ipertensione di grado 3 o peggiore. Il 30 per cento delle pazienti ha sviluppato una proteinuria di grado uno o due, di cui la metà entro le due settimane e le rimanenti in 6, riconoscendo come unico fattore di rischio significativo la dose iniziale di cediranib.
Interessante è stato il rilievo che fino al 65% delle pazienti con ipertensione di grado 3 o peggiore non ha sviluppato la proteinuria e che, almeno in una paziente, la proteinuria si è sviluppata prima dell’ipertensione. Tali dati hanno suggerito che questa classe d’inibitori del recettore VEGF possano produrre due effetti collaterali attraverso differenti meccanismi. Peraltro, l'insorgenza precoce dell’ipertensione potrebbe risultare dall’inibizione acuta della vasodilatazione mediata dal VEGF. Tali dati portano a stimolare, soprattutto nei pazienti più anziani, attenzione e vigilanza nella diagnosi precoce e nella gestione delle tossicità di questa classe di farmaci in continua espansione.
Rischio d’ipotensione grave con l’uso di CCB/macrolidi
Alissa J Wright e collaboratori dell’University of Toronto, ON hanno eseguito il primo tentativo rigoroso per descrivere le conseguenze cliniche dell’interazione tra CCB (calcium-channel blockers) e antibiotici macrolidi (CMAJ 2011; DOI:10.1503/cmaj.100702). Nella loro popolazione di studio hanno analizzato circa un milione d’individui d’età superiore ai 65 anni, trattati con un singolo BCC tra il 1994 e il 2009. Di questi pazienti 7100 erano stati ricoverati in ospedale per ipotensione o shock e 176 avevano ricevuto un antibiotico macrolide. In particolare, 36 avevano usato eritromicina, 100 claritromicina e 40 azitromicina in un intervallo di sette giorni immediatamente prima del ricovero o del controllo. I ricercatori hanno, così, trovato una forte associazione tra l'uso di eritromicina e i ricoveri ospedalieri per ipotensione con un rischio quasi sei volte maggiore di bassa pressione arteriosa (odds ratio 5.8) e un rischio più basso, ma pur sempre significativo, associato all'uso di claritromicina (OR 3.7).Non si dimostrava, invece, alcun nesso di tal genere con l'utilizzo dell’azitromicina (OR 1,5). Questo effetto può trarre origine dal fatto che farmacologicamente i macrolidi, inibendo il citocromo P450, che metabolizza i CCB, determinerebbero il loro accumulo aumentandone la potenziale tossicità. Difatti, l’azitromicina, a differenza degli altri composti della stessa classe, non possiede queste proprietà. I medici devono, quindi, mostrare cautela e attenzione nell’uso di questi farmaci per evitare spiacevoli danni ai loro pazienti.
Steroidi e dopamina fatali nello shock settico?
Daniel De Backer dell’University Hospital Erasme a Bruxelles in Belgio e collaboratori hanno segnalato un loro studio alla Society of Critical Care Medicine 40th Critical Care Congress il 25 gennaio 2011 a San Diego, California in cui si dimostrava che i pazienti con shock, in particolare quello settico, trattati con steroidi e dopamina dimostravano un aumento della mortalità a 28 giorni, pari al 56% rispetto al 49% di quelli randomizzati a noradrenalina. Nei casi, invece, senza steroidi, la mortalità a 28 giorni era in sostanza la stessa, al 49%.La ricerca è conseguita, in via secondaria, allo studio SOAP (Sepsis Occurrence in Acutely Ill Patients),condotto sempre da De Backer e collaboratori (N Engl J Med. 2010,362:779-789). In effetti, sia la dopamina sia la noradrenalina sono farmaci vasopressori di prima linea, raccomandati nel trattamento dello shock. Il SOAP è stato intrapreso per stabilire se un agente si proponeva, in via di superiorità, agli altri nel ripristinare e mantenere la pressione sanguigna. A tal fine 858 pazienti con shock sono stati arruolati a dopamina e 821 a noradrenalina. La dopamina, invero, si è associata a un aumento non significativo di mortalità a 28 giorni, nei confronti della noradrenalina (52,5% vs 48,5%; odds ratio [OR], 1.17, 95% intervallo di confidenza [IC], 0,97-1,42, p = 0,10). La dopamina, però, si è anche associata significativamente a maggiori eventi aritmici. Si sono, difatti, riscontrati 207 eventi (tasso 24,1%) con dopamina contro i 102 (tasso 12,4%) con noradrenalina (P <.001). L’analisi dei dati, con particolare osservazione sull'uso del prednisolone e dell’idrocortisone entro 24 ore dalla randomizzazione alla dopamina e la noradrenalina, ha permesso di appurare i risultati in precedenza espressi. In conformità a questi risultati, gli AA sconsigliano l’uso prolungato di dopamina come vasopressore.
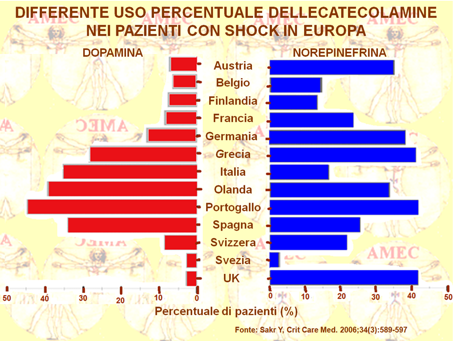
In un precedente lavoro di ricerca Sakr Y del Department of Intensive Care, Erasme Hospital, Free University of Brussels, Belgium.e i suoi collaboratori (Crit Care Med. 2006;34(3):589-597), sempre con l’obiettivo di chiarire il controverso supporto ottimale dei farmaci adrenergici nello shock, hanno indagato sull’influenza della dopamina in tale campo con uno studio di coorte, multicentrico, osservazionale in collaborazione di 198unità europee di terapia intensiva.Lo shock è stato definito come una compromissione emodinamica con richiesta di somministrazione di catecolamine vasopressorie.
Dei 3.147 pazienti, 1.058, il 33,6%, erano in shockin qualsiasi momento e 462, il 14,7%, nella variante settica. Il tasso di mortalità per lo shock è stato 38,3% contro il 47,4% per quello settico. Il 35,4%, corrispondente a 375 pazienti, ha ricevuto dopamina e il 64,6%, 683 in numero, non l’ha mai ottenuta. L’età, il sesso, il Simplified Acute Physiology Score II e il Sequential Organ Failure Assessment score erano comparabili tra i due gruppi. Il gruppo di dopamina aveva dimostrato valori più alti di terapia intensiva (42,9% vs 35,7%, p = .02) e tassi di mortalità ospedaliera (49,9% vs 41,7%, p = .01). La curva di sopravvivenza di Kaplan-Meier ha mostrato una ridotta sopravvivenza a 30 giorni nel gruppo di dopamina (log rank p = 4.6, = 0,032). In un'analisi multivariata, con i risultati dell’unità di terapia intensiva come fattore indipendente, l'età, il cancro, i ricoveri medici, la media maggiore del Sequential Organ Failure Assessment score, il maggiore equilibrio medio dei fluidi e la somministrazione di dopamina sono risultati fattori di rischio indipendenti per la mortalità da shock. Lo studio osservazionale suggerisce, invero, che la somministrazione di dopamina può essere associata a tassi di aumento della mortalità in stato di shock.
Attenzione nel trattare i crampi alle gambe con il chinino
La FDA ha annunciato il pericolo che può derivare dal prescrivere il chinino (qualaquin) per i crampi notturni alle gambe perché si possono causare effetti ematologici gravi e negativi per la vita. Il farmaco, in effetti, è commercializzato dall’AR Scientifica e approvato solo per la malaria non complicata, causata dal parassita Plasmodium falciparum. La terapia può determinare trombocitopenia con gravi emorragie o sindrome emolitico-uremica/porpora trombotica trombocitopenica, con possibili danni permanenti ai reni.La FDA ha annunciato di aver ricevuto 38 segnalazioni di gravi eventi avversi negli Stati Uniti, attraverso il suo Adverse Event Reporting System, associati alla somministrazione di chinino tra l’aprile 2005 e il primo ottobre 2008. Di queste, due riportavano il decesso dei pazienti e solo in una l’assunzione del farmaco rispondeva alla cura della malaria.
Ritiro dal mercato della sibutramina
La FDA ha annunciato l’8 ottobre 2010 il ritiro dal mercato, da parte dell’Abbott Laboratories, della sibutramina (meridia), farmaco per curare l’obesità, alla luce dei risultati clinici di studio post-marketing che hanno rivelato un aumentato rischio d’infarto miocardico e d’ictus. Lo SCOUT (Sibutramine Cardiovascular Outcomes Trial) ha dimostrato, in effetti, in una coorte di pazienti in trattamento con sibutramina, confrontato con un gruppo placebo,un aumento del 16% del rischio d'eventi cardiovascolari gravi, come l’infarto miocardico e l’ictus non fatali, la necessità di rianimazione per arresto cardiaco e la morte. Dopo la votazione consultiva, la FDA ha concluso che i rischi cardiovascolari posti dalla Sibutramina, approvata nel 1997, erano superiori ai vantaggi. John Jenkins, peraltro, direttore dell’Office of New Drugs at the FDA's Center for Drug Evaluation and Research,comunicava che circa 100.000 statunitensi erano in corso di cura con la sibutramina. La raccomandazione, comunque, della FDA ai medici, di interrompere la prescrizione di sibutramina e ai pazienti di sospendere l'assunzione e di parlare con il loro medico circa i regimi alternativi di perdita di peso, era seguita dalla dichiarazione di non conoscenza di una possibile sindrome da astinenza. Gli eventi cardiovascolari, osservati nello SCOUT, deriverebbero, probabilmente, dall’aumento della pressione sanguigna e della frequenza cardiaca, legate alla sibutramina, che scompaiono con la sospensione del farmaco.Farmaci disponibili, in alternativa a breve termine per la perdita di peso, includono, a ogni buon conto, la fentermina e il dietilpropione. L’orlistat, peraltro, è disponibile sia in una forma over-the-counter (Alli, GlaxoSmithKline) sia convenzionale (xenical, Hoffman-La Roche). A tal proposito, la FDA lo scorso26 maggio 2010 ha rivisitato l'etichetta per entrambe le versioni di orlistat per mettere in guardia sui rari casi di grave danno epatico, associato al suo uso.L'annuncio è derivato da una revisione completa di tutti i dati disponibili, provenienti da studi preclinici, clinici, post-marketing dall’aprile 1999 al 7 agosto 2009. La FDA ha, così, identificato 13 casi post-marketing di danno epatico grave, di cui 12 casi, non degli USA, che hanno coinvolto xenical,senza aver ancora stabilito una relazione causale. Alcuni pazienti sono morti o hanno avuto bisogno di un trapianto di fegato.
Thelin (sitaxentan sodium) e grave danno epatico
La Pfizer ha annunciato di ritirare volontariamente thelin dalla commercializzazione mondiale il 10 dicembre 2010, a causa di segnalazioni di grave danno epatico con la somministrazione del prodotto. Su tale base, i pazienti in terapia con il farmaco devono essere avvertiti di usare al più presto principi attivi alternativi, consultando il proprio medico di fiducia senza sospendere autonomamente il trattamento. Essorimane, però, disponibile sul mercato durante un periodo di transizione senza essere, in ogni caso, prescritto a nuovi pazienti. Peraltro, sono interrotti tutti gli studi clinici che lo interessano. Thelin,il cui principio attivo è il sitaxentan, è stato autorizzato nell’Unione Europea (EU) nel 2006 per il trattamento dell’ipertensione arteriosa polmonare. Pfizer ha annunciato che il provvedimento è consequenziale a una revisione dei casi fatali associati a grave danno epatico, che hanno compreso un caso del 2009 nel Regno Unito e un altro in India e un altro ancora in Ucraina nel 2010. È stata ipotizzata anche un’idiosincrasia. Comunque, sulla base delle conoscenze attuali e in considerazione della disponibilità di trattamenti alternativi, si è concluso che complessivamente i benefici clinici di thelin non superino i rischi nei pazienti affetti da ipertensione arteriosa polmonare. L'AIFA, dal suo canto, ha voluto ribadire l'importanza della segnalazione da parte della classe medica delle sospette reazioni avverse da farmaci, quale strumento indispensabile per confermare un favorevole rapporto beneficio/rischio nelle loro reali condizioni d’impiego.
Lorcaserin respinto dalla FDA per problemi di sicurezza
Alla fine dell'anno scorso, la FDA ha respinto una domanda di uso del lorcaserin, un farmaco per perdere peso, per il fatto che i suoi benefici si erano dimostrati sorpassati dai problemi di sicurezza. La decisione è stata motivata da studi di cancerogenicità sugli animali, ma anche dagli effetti collaterali derivati di studi clinici randomizzati. Sulla base che farmaci con un meccanismo d'azione simile sono stati riconosciuti causa di aumentato rischio di malattie delle valvole cardiache, sono stati di proposito progettati trial per verificare la stessa tossicità del lorcaserin.
Il governo francese boccia la pillola dimagrante mediator
Xavier Bertrand, ministro della sanità francese,il gennaio 2011, ha promesso di accelerare i controlli di sicurezza su decine di medicinali e di dare uno scossone alle procedure di autorizzazione di messa in commercio dei farmaci, dopo un rapporto schiacciante su una pillola per il controllo del peso, il mediator. La società francese Servier ha venduto a circa 5 milioni di persone, diabetici e/o sotto dieta, il prodotto tra il 1976 e il novembre 2009, data del suo ritiro in Francia. Il suo principio attivo è il benfluorex, peraltro, con dubbi sulla sua efficacia, emersi già alla fine del 1990. Il benfluorex, derivato funzionale della fenfluramina, che riduce l'assorbimento intestinale dei grassi per inibizione delle lipasi pancreatiche, riduce anche l'iperglicemia, migliorando l'utilizzo periferico del glucosio. Da notare che in Italia il prodotto è stato commercializzato fino a tutto il 2003 con la denominazione di mediaxal e l'indicazione della cura delle forme iperdislipidemiche, resistenti alla sola dieta. È stato anche consigliato come coadiuvante nell'obesità, associata ad alterazioni del metabolismo glico-lipidico, ma poi ritirato dalla stessa ditta produttrice. In Spagna l'analoga specialità è stata ritirata nel marzo 2003 per la segnalazione di effetti avversi, cardiovascolari gravi, comprendenti l’ipertensione polmonare e le valvulopatie, similari a quelli di farmaci a essa strutturalmente correlati, quali la fenfluramina e la dexfenfluramina. L’AFSSAPS (French Agency for Safety of Health Products) ha in quest’occasione annunciato che sull’esposizione totale di sette milioni di persone-anno, tra il 1979 e il 2009, la stima dei decessi per sua causa è stata di circa 500 persone. Nel periodo di follow-up dei 303.000 pazienti monitorizzati, di cui 73% donne di età media di 53 anni, 597 sono stati ospedalizzati per malattia valvolare, il 50% ha subito intervento chirurgico valvolare e 64 sono morti, di cui 33 dopo l'intervento. Dei decessi, 46 sono stati considerati attribuibili alla malattia valvolare,soprattutto mitralica e aortica. Peraltro, tra il 2006 e il 2009, 556 pazienti sono stati ospedalizzati per insufficienza valvolare con un tasso di 184 per 100 000, 46 per 100 000 persone-anno, 303, il 54%, per insufficienza mitralica, 270, il 48 %, per insufficienza aortica, 77, il 18%, per il rigurgito della tricuspide e 41 per patologia multi valvolare. Delle oltre 303 000 persone esposte al benfluorex nel 2006, 99 sono state ricoverate con diagnosi d’ipertensione polmonare o con una condizione a essa connessa. In caso d’ipertensione polmonare preesistente, il farmaco ha dimostrato di peggiorare lo stato clinico.
Divieto di utilizzo di primene
L’AIFA (Agenzia Italiana del Farmaco) ha comunicato il divieto di utilizzo di primene 20 fl 100 ml 10% (AIC 02690599) a causa di concentrazioni di sodio cloruro riportate in etichetta e in RCP non compatibili con le indicazioni e la formulazione autorizzate. Si precisa anche che solo in casi di assoluta necessità e urgenza e in mancanza di alternativa, tale medicinale potrà essere praticato. Bisogna, però, ricordare che sull'etichetta e sul RCP è erroneamente riportata una quantità di sodio non presente nel prodotto, mentre i cloruri sono pari a diciannove mMol/Litro e non a 15,6 mMol/ml.
Pertanto, non permettendo la legislazione italiana l'acquisto e la successiva spedizione di "farmaci" via internet o per corrispondenza, prodotti di tal genere sono prenotabili solo dopo aver verificato la reale disponibilità e la possibilità di vendita al pubblico.
Corticosteroidi per via inalatoria e rischio di diabete
Suissa S e collaboratori del Center for Clinical Epidemiology, Lady Davis Research Institute, McGill University, Montreal, sulla base della scarsa conoscenza dicorrelazione tra corticosteroidi ad alte dosi per via inalatoria e insorgenza di diabete, utilizzando il database di assicurazione sanitaria del Quebec, dopo aver identificato una coorte di pazienti trattati dal 1990 al 2005 per malattie respiratorie, hanno stimato i rapporti tra tasso d’insorgenza e progressione della malattia con l'impiego corrente di corticosteroidi per via inalatoria, usando un’analisi caso-controllo con aggiustamento per età, sesso, gravità della malattia respiratoria e le condizioni di comorbidità.I pazienti sono stati seguiti fino al 2007 o fino all'insorgenza di diabete (J Am Med. 2010; 123:1001-1006.). Una sottocorte in trattamento con farmaci ipoglicemizzanti è stata seguita fino alla progressione della malattia.Dei 388.584 pazienti, 30.167 hanno sviluppato di diabete in 5,5 anni di follow-up (tasso d’incidenza, 14.2/1000/anno), e 2.099 pazienti sono stati successivamente passati dalla terapia con ipoglicemizzanti orali a quella insulinica (tasso d’incidenza, 19.8/1000 / anno).I partecipanti con uso corrente di corticosteroidi per via inalatoria hanno avuto un aumento del 34% del tasso di diabete (Rate Ratio (RR), 1,34; intervallo di confidenza 95% [IC], 1,29-1,39) e del tasso di progressione del diabete (RR, 1,34; 95% CI, 1,17-1,53). Il più alto dosaggio di corticosteroide per via inalatoria, equivalente ad almeno 1000 μg/die difluticasone, si è associato agli aumenti maggiori di rischio (RR 1,64 [95% CI, 1,52-1,76] per il tasso di diabete, RR 1,54 [95% CI, 1,18-2,02] per la progressione del diabete)rispettivamente rispetto al non uso.In conclusione, lo studio ha dimostrato che nei pazienti con malattie respiratorie l'uso di corticosteroidi inalatori si associa a un modesto aumento di rischio di diabete e di progressione della malattia. Il rischio è più pronunciato alle dosi più elevate del farmaco, che deve essere più attentamente valutato sul piano del rischio-beneficio. Ne consegue che i medici, prima di iniziare una terapia con alte dosi di corticosteroidi per via inalatoria, dovrebbero considerare un possibile errore del ricambio glicidico dei loro pazienti e riservare il trattamento solo alle situazioni in cui il beneficio è evidente. Gli autori rilevano, infatti, che, sebbene questi farmaci siano consigliati solo per i pazienti con BPCO più grave, la prassi attuale è quella della prescrizione per oltre il 70% dei casi, inclusi quelli con malattia meno grave. Tale dato, unito all’aumento della prevalenza sia della BPCO sia del diabete con il progressivo invecchiamento demografico della popolazione, conduce a serie preoccupazioni per il futuro.
Attenzione all’'uso della terbutalina nel trattamento del parto pretermine
Il 17 febbraio 2011 la FDA ha comunicato che la terbutalina iniettabile non deve essere usata nelle gravide, sia in ospedale sia ambulatorialmente, nella prevenzione o nel trattamento prolungato (oltre 48-72 ore) del parto pretermine, a causa del rischio potenziale di gravi problemi al cuore materno e di morte. Tale avvertimento, peraltro, era riferibile anche alla somministrazione orale per la non provata efficacia del farmaco e per problemi di sicurezza simili alla forma iniettabile. Invero, questo principio attivo, primieramente approvato per prevenire e curare l’asma da broncospasmo, associato a bronchite ed enfisema, è a volte utilizzato off-label (un uso non approvato) per condizioni di acuzie in ostetricia, compresi il trattamento del parto pretermine e quello dell’iperstimolazione uterina. La terbutalina, comunque, è stata utilizzata anche off-label per lunghi periodi, nel tentativo di evitare ricorrenti parti pretermine.
È bene che le pazienti sappiano:
- devono essere consapevoli del fatto che gli effetti collaterali gravi, tra cui problemi di cuore della madre e di morte, sono stati dimostrati dopo un uso prolungato di terbutalina per gestire il parto pretermine,
- ci sono situazioni gravi in cui un medico può decidere che l'uso a breve termine di terbutalina iniettabile in ambito ospedaliero può essere vantaggioso in una donna incinta,
- la terbutalina orale non deve essere usata per il trattamento di parto pretermine o per prevenire recidive di parto pretermine,
- se si sta usando terbutalina per un'altra condizione medica (ad esempio, asma), parlarne con il proprio medico se si è in gravidanza,
- si devono segnalare eventuali effetti collaterali durante l’uso di terbutalina per via orale o iniettabile.
È bene che i medici sappiano:
- devono essere consapevoli del fatto che la morte e gravi reazioni avverse, tra cui aumento del battito cardiaco, l’iperglicemia transitoria, l’ipopotassiemia, le aritmie cardiache, l’edema polmonare e l'ischemia del miocardio sono stati dimostrati dopo la somministrazione prolungata di terbutalina per via orale o iniettabile in donne in gravidanza.
- Il trattamento con terbutalina iniettabile anche con infusione continua di pompa non deve essere usato oltre le 48 - 72 ore. In particolare, la terbutalina iniettabile non deve essere usata in ambito ambulatoriale o a domicilio.
- Vi sono alcune condizioni ostetriche, in cui il personale sanitario può decidere che il beneficio d’iniezione di terbutalina per una singola paziente in ambiente ospedaliero superino chiaramente i rischi.
- La terbutalina orale è, comunque, controindicata per il trattamento o la prevenzione del parto pretermine.
- Le relazioni con gli eventi avversi che coinvolgono la terbutalina al programma FDA MedWatch sono consultabili utilizzando le informazioni nel box "Contatti" (National Asthma Education and Prevention Program (NAEPP). Working Group Report on Managing Asthma During Pregnancy: Recommendations for Pharmacologic Treatment—Update 2004. NIH Publication No. 05-5236. Bethesda, MD: U.S. Department of Health and Human Services; National Institutes of Health; National Heart, Lung, and Blood Institute, 2004. Available from:
http://www.nhlbi.nih.gov/health/prof/lung/asthma/astpreg/astpreg_full.pdf1. Accessed November 19, 2010).
Uso del valproato nella gestante e gravi difetti nel nascituro
Katherine L. Wisner e collaboratori del Western Psychiatric Institute and Clinic, Pittsburgh, Pennsylvania hanno studiato l’uso del valproato in 40.526 individui come stabilizzatore non antipsicotico dell'umore rilevandolo il più usato con il 32,3%, seguito dal gabapentin (26,4%), dalla lamotrigina (16,7%), dal topiramato (13.%), dal litio (13,0%), dall’oxcarbazepina (5,1%) e dalla carbamazepina (3,2%). Peraltro, il 23,4% delle prescrizioni di valproato si riferiva a giovani donne, pur essendo meno propense degli uomini e delle donne anziane a utilizzarlo. Tale dato, unito alla nota tossicità riproduttiva del farmaco, conduce a importanti considerazioni e perplessità che devono stimolare un'opportuna prevenzione. Infatti, tale principio attivo, ampiamente usato per trattare il disturbo bipolare, ha dimostrato di aumentare il rischio di sviluppo della sindrome dell'ovaio policistico e dei difetti alla nascita. Peraltro, lo studio NEAD (Neurodevelopmental Effects of Antiepileptic Drugs)hadimostrato che l'esposizione prenatale al farmaco aumenta il rischio di deficit cognitivi a lungo termine nella prole(NeurologyNovember 30, 2010 75:1948-1949). Peraltro, oltre al deterioramento cognitivo, si sono ribaditi tassi di gravi eventi avversi più alti, rispetto ad altri anticonvulsivi, incluse le malformazioni congenite e la morte fetale.Per tali motivi nel 2009 l’US Food and Drug Administration ha emesso un avviso di allerta sulla sicurezza del farmaco nei neonati per il rischio di difetti del tubo neurale, difetti cranio-facciali e malformazioni cardiovascolari.
Difetti di nascita con l’uso del topiramato
Secondo nuovi dati del North American Antiepileptic Drug Pregnancy Registry, il topiramato (Topamax, Ortho-McNeil Janssen) usato da solo nel primo trimestre di gravidanza esporrebbe i neonati all’1,4% di schisi orali rispetto allo 0,38- 0,55% degli altri farmaci antiepilettici. Peraltro, ilt asso di prevalenza è solo dello 0,07% nei nati da madri che non hanno avuto l'epilessia e non sono state, quindi, trattate con alcun antiepilettico. Questi risultati hanno spinto l'Agenzia a rafforzare gli avvertimenti sull'etichetta del farmaco, modificando la stessa classificazione di gravidanza nella categoria “D” dalla “C “, più bassa per l'assenza di rischio nell’uomo. Ciò significa che ci sono dati che mostrano evidenze positive di rischio fetale, tenendo sempre conto, però, che in alcune situazioni di gravidanza i benefici del farmaco possono superare i rischi. Di conseguenza, ne deriva la raccomandazione ai medici dimettere in guardia le donne in età fertile circa la possibilità del rischio di tali difetti alla nascita in caso di trattamento con il farmaco durante la gravidanza. Il topiramato è un anticonvulsivante, approvato per il trattamento di alcune, determinate crisi epilettiche e per prevenire l'emicrania. Pur tuttavia, non è indicato per il trattamento del mal di testa generico. Il farmaco è usato anche in forma off-label per il trattamento della perdita di peso, per la dipendenza da alcol e malattie psichiatriche, come il disturbo bipolare. Dal suo canto,
Kim Meador dell’University of Florida in Gainesvilleha diretto il NEAD (Neurodevelopmental Effects of Antiepileptic Drugs, ampio studio osservazionale prospettico su più di 300 donne epilettiche di 25 centri negli Stati Uniti e nel Regno Unito (Neurology2008;71:272-276). Gli autori, premettendo che è ampiamente riconosciuto che l'esposizione prenatale ai farmaci antiepilettici più antichi aumenta il rischio di MCM (major congenital malformations)nella popolazione generale dall’1% - 2% al 4% - 9%, hanno ribadito che poco si sa circa i rischi con alcuni dei più recenti agenti, tra cui il vigabatrin, il gabapentin, la zonisamide, la tiagabina, il pregabalin e il topiramato. Quest’ultimo, peraltro, si è dimostrato teratogeno negli animali. Il NEADha incluso donne con epilessia rimaste incinte durante il trattamento con topiramato sia in monoterapia sia in combinazione con altri farmaci antiepilettici, in particolare il valproato, che si è dimostrato in genere associato a scarse malformazioni anatomiche ed esiti cognitivi nella prole. L'outcome principale è stato il tasso di MCM. Gli end point secondari comprendevano il rischio di specifiche MCM, il tasso di malformazioni minori, il peso alla nascita e il periodo gestazionale al momento del parto. Corrispondenti alle 203 gravidanze si sono contati 178 nati vivi, di cui31 con un'anomalia di qualche tipo e 16 con un MCM, tre dei quali in donne in monoterapia con topiramato e il resto in politerapia. Gli investigatori non hanno trovato un’apparente associazione dose-risposta sia con le esposizioni in monoterapia sia in politerapia.
Nota informativa AIFA sulle fiale di vitamina “C SALF”
L’Agenzia Italiana del Farmaco, insieme a S.A.L.F. S.p.A. ha emesso nel marzo 2011 una nota informativa sulla possibile sovrapressione all’interno delle fiale di vitamina “C SALF” con loro conseguente rottura. Ne deriva la raccomandazione di conservare il medicinale a temperature inferiori ai 25° C e al riparo dalla luce e di attenersi alle istruzioni fornite in caso di ferite dell’operatore per la frammentazione del vetro.
Comunque, già nelle istruzioni d’uso è ribadito che “A volte si può verificare che all’interno della fiala vi sia sovrapressione: per questo si consiglia di aprirla proteggendo le dita con un tampone”.
Triplice combinazione antipertensiva in USA
Il 27 dicembre 2010la FDA statunitense ha approvato la commercializzazione della prima combinazione di una triplice terapia antipertensiva contenente aliskiren, inibitore diretto della renina, l’amlodipina, calcio-antagonista e l'idroclorotiazide, diuretico.
Robot per testare la tossicità dei prodotti chimici
Il 10 marzo 2011 il National Institutes of Health e diverse altre agenzie federali hanno presentato un nuovo sistema automatico per lo screening ad alta velocità della potenziale tossicità di diverse sostanze chimiche, segnando l'inizio della Tox21, nuova fase di collaborazione per la tutela della salute umana. Il sistema di robot si trova presso il Centro di Genomica Chemical NIH (NCGC) a Rockville, Maryland e si pone l’obiettivo di sviluppare metodi per predire, in modo più efficace, gli effetti dannosi delle sostanze chimiche sulla salute umana e sull'ambiente.Allo stato attuale sono stati controllati 10.000 prodotti chimici di articoli industriali e di consumo, additivi alimentari e farmaci, dopo A thorough analysis and prioritization process from more than 200 public databases of chemicals and drugs used in the United States and abroad was conducted to select the initial 10,000 chemicals for testing.un'analisi approfondita e un processo di definizione delle priorità, derivato dalla consultazione di dati di più di 200 banche pubbliche di prodotti chimici e farmaci utilizzati negli Stati Uniti e nel mondo. In tal modo, ci si prefigge di ottenere una migliore comprensione tossicologica con la capacità di analisi migliori e più intelligenti delle sostanze chimiche e di evidenze più veloci rispetto a quanto è dato con i sistemi sino ad ora adottati. Gli organi precostituiti potranno, quindi, essere in grado di fornire più rapidamente le informazioni ai decisori sanitari e normativi e altri sulle sostanze potenzialmente pericolose, in modo che si possano prendere decisioni consapevoli e tempestive per tutelare la salute pubblica. Difatti, la comprensione delle basi molecolari del rischio tossicologico è senza alcun dubbio fondamentale per la protezione della salute umana e dell'ambiente.
"Tox21 has used robots to screen chemicals since 2008, but this new robotic system is dedicated to screening a much larger compound library," said NHGRI Director Eric Green, MD, Ph.D.
The 10,000 chemicals screened by the robot system include compounds found in industrial and consumer products, food additives, and drugs.allo stato attuale sono stati testatiaa in tal modoi