







| Indice |
|---|
| notiziario gennaio 2011 N°1 - DANNI DA FARMACI |
| I danni da farmaci |
| Farmacologia istituzionale e medicina alternativa e complementare |
| ADR nell’uso della CAM in bambini |
| Tutte le pagine |
I danni da farmaci
Il problema del danno da farmaci e della sicurezza delle terapie costituisce una priorità sempre più attuale e impellente. Al momento, nel ciclo produttivo e distributivo, sia da parte della comunità scientifica sia da parte dell’autorità regolatoria, è molto sentito il bisogno della valutazione e della pubblicizzazione del rapporto rischio/beneficio del farmaco, in modo organizzato, trasparente e integrato.
Grande importanza assume, pertanto, un progetto funzionale di risk management che deve iniziare con un programma di analisi di farmacovigilanza focalizzata sul rischio, rivolta a colmare le conoscenze ineluttabilmente presenti, specialmente nelle prime fasi di commercializzazione di un prodotto. Per l’appunto, una strategia per minimizzare il rischio risiede nel rilievo e nell’integrazione dei dati in maniera incrementale, giacché il livello di sicurezza di una sostanza è intrinseco e immutabile. Peraltro, non sono nuovi gli strumenti idonei al ricavo dei dati e alla valutazione del rischio stesso. Pertanto, il valore della fonte dei risultati, in funzione del loro corrispettivo potere predittivo, assume, nell’ambito delle specifiche categorie di esso, particolare importanza nella stima del rischio.
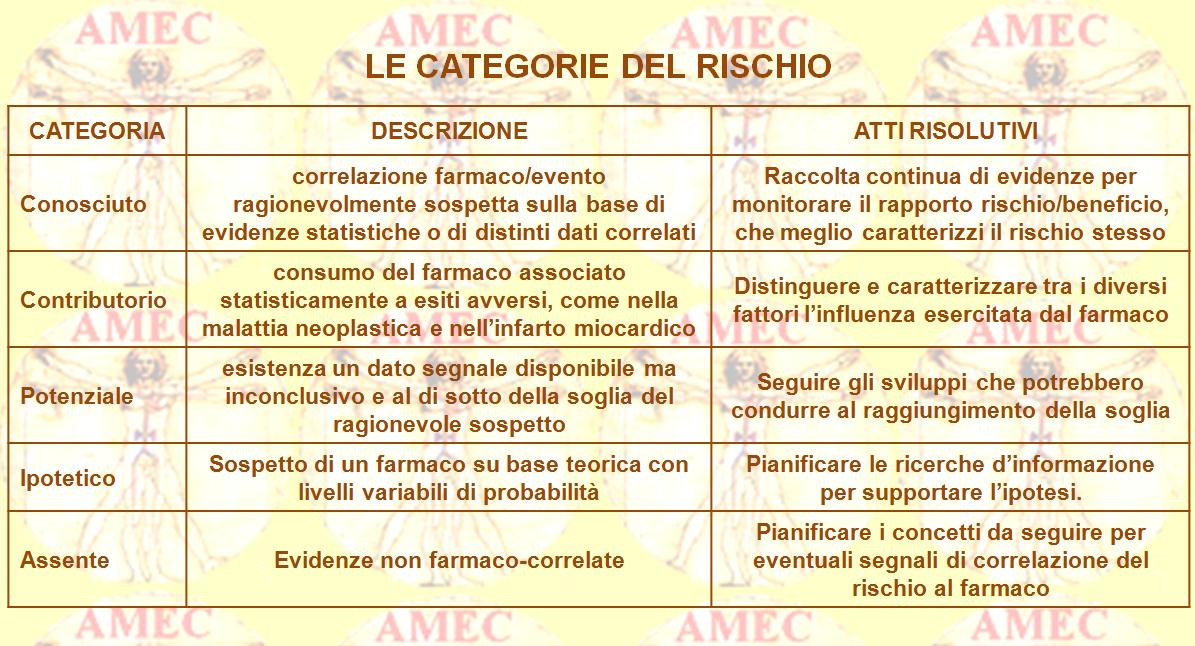
Suddividendo, così, le categorie del rischio e indirizzando i dati sotto ciascuna di esse, si può, di conseguenza, progettare un piano di Risk Managment trasparente, integrato nel ciclo produttivo e di utilizzo del farmaco, in grado di mantenere una rivalutazione continua del rapporto Rischio/Beneficio.
In quest’ultimo processo, una corretta strategia deve iniziare con la stima delle proprietà biologiche, chimico-fisiche del prodotto per passare poi ad altri parametri, quali la frequenza e la gravità degli eventi dannosi causati, l’analisi delle variabili e come queste incidono sul suo profilo primario.
Non di ultimo interesse è l’impatto del rischio sul piano terapeutico e come esso si bilancia con la valutazione del benessere e con l’implementazione di misure di risk mitigation per posizionare il prodotto nei maggiori margini di sicurezza terapeutica.

Tutto ciò deve derivare dalla:
- causalità o meno di un’ADR,
- frequenza/gravità di essain una data popolazione,
- variabilità del rischio, come comorbidità e/o uso di altri farmaci,
- impatto sul trattamento o sul benessere,
-
valutazione del rapporto rischio/beneficio e della risk mitigation
per il migliore posizionamento del farmaco.
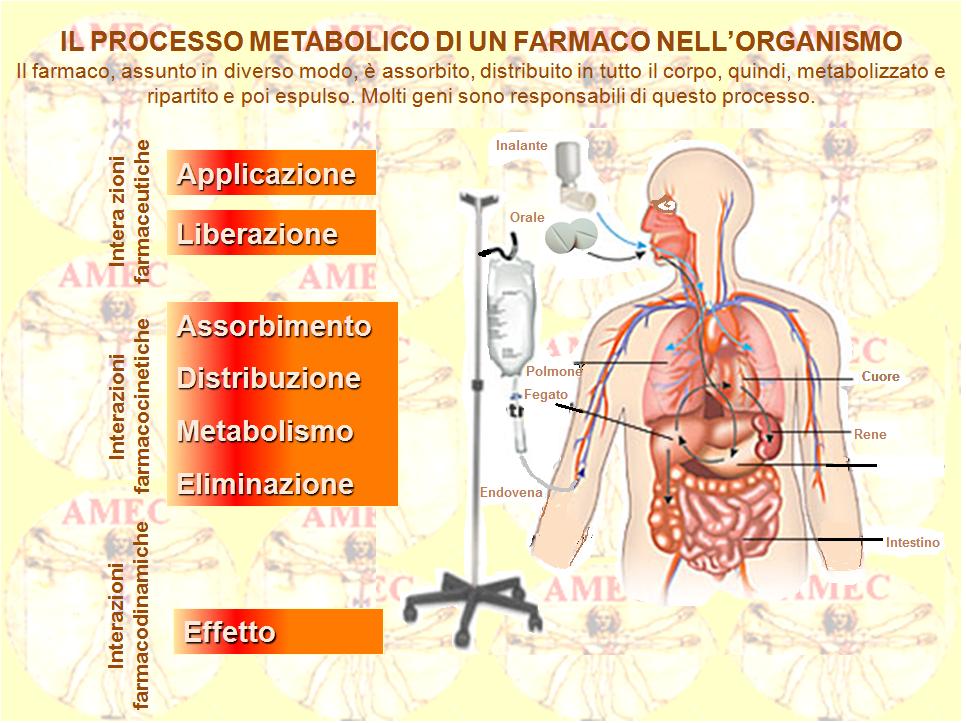
Com’è noto, ogni farmaco ha un suo destino nel nostro organismo:
- viene assorbito e metabolizzato in un certo modo,
- circola nel torrente sanguigno in altri modi, penetrando solo in quelle cellule provviste di particolari recettori, funzionanti come serrature molecolari,
- viene, quindi, agganciato,
- ha, pertanto, particolari bersagli in questo o quell'organo nel quale svolge la sua azione terapeutica,
- viene eliminato in diversi altri modi.
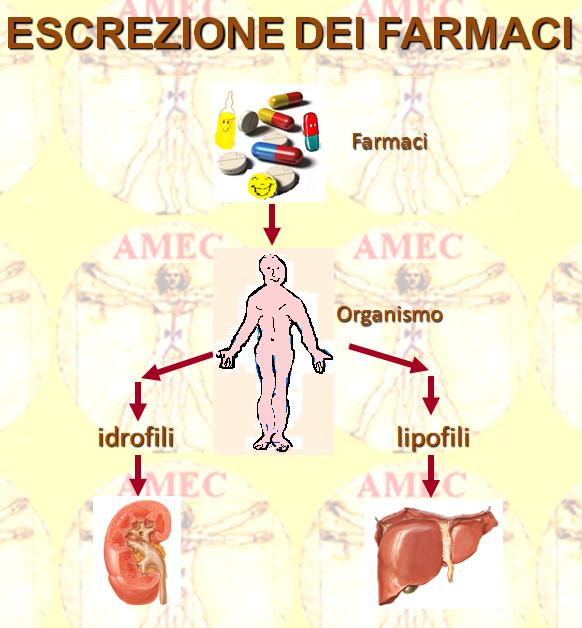
Di solito, il farmaco blocca i recettori cellulari ostacolandone la funzione, mentre in altra condizione entra nella cellula e ne modula l’attività o la riproduzione. Infine, dopo un tempo variabile da farmaco a farmaco, avviene la sua eliminazione locale nella cellula oppure per opera di altri tessuti, come il fegato e/o i reni. Peraltro, in rapporto alle diverse circostanze, è possibile misurare la concentrazione sanguigna o urinaria del prodotto chimico, traendo nozioni che permettono di spiegare da una parte la mancata efficacia dovuta a insufficiente assorbimento e dall’altra l’effetto tossico per le sue quantità eccessive.
Un farmaco, comunque, somministrato per un effetto terapeutico, può promuovere nel nostro organismo numerose conseguenze con effetti indesiderati, sotto la variante più lieve di effetti collaterali o sotto forma di una reazione allergica. In tutti i casi, la distinzione tra effetto curativo/indesiderato è in qualche modo discutibile perché il curativo di oggi potrebbe sempre divenire l'effetto cercato in futuro. Gli antistaminici, ad esempio, con effetto collaterale di sonnolenza, sono stati poi usati come sedativi e così pure l’aspirina ha trovato ampia diffusione d’uso nella prevenzione della malattia cardiovascolare per i suoi effetti collaterali sull’aggregazione piastrinica.
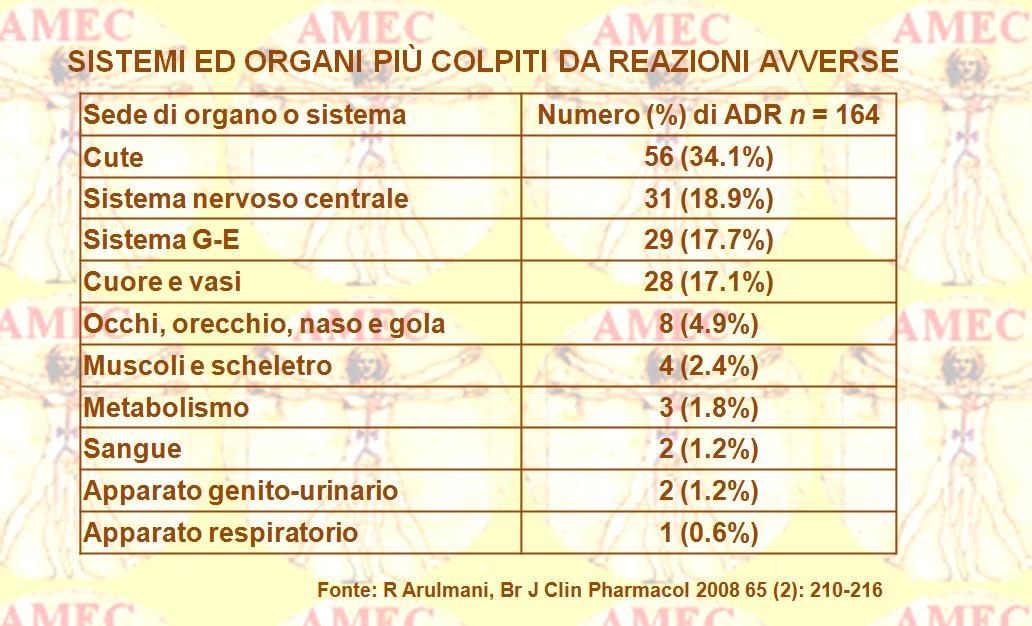
Negli ultimi quindici anni, molti farmaci, appartenenti a diversi gruppi terapeutici, sono stati ritirati dal commercio per ragioni di sicurezza. Questo è stato il risultato di effetti collaterali, conseguenze prevedibili e indesiderabili dose-dipendenti durante un piano terapeutico. In altri casi si è dimostrata una tossicità da sovradosaggio, prevedibile per dosi sopra il range terapeutico per un determinato paziente. In quest’occasione, invero, vi può essere una sovrapposizione con la precedente forma, soprattutto nell’uso di agenti con limitato indice terapeutico. Peraltro, la gravità del quadro clinico, spesso determinata dalle malattie di base, come la grave insufficienza epatica e/o renale, è abitualmente dose-dipendente.
Le allergie ai farmaci, invece, non dose-dipendenti, sono abbastanza frequenti e ricorrono in casi di alterata reattività dopo un precedente uso del farmaco, che assume, quindi, il ruolo di antigene o allergene. Il quadro clinico, indipendente dalle proprietà farmacologiche della sostanza, è dominato dalla reazione antigene-anticorpo. Pur tuttavia, con un’attenta e diligente anamnesi, seguita da test appropriati, si possono riconoscere i casi a rischio, tanto da poter considerare le allergie sufficientemente prevedibili e prevenibili.
Le reazioni avverse inaspettate e peculiari, ricorrenti in una piccola percentuale d’individui, chiaramente non di natura allergica,non legate alle proprietà farmacologiche, definiscono, invece, l’idiosincrasia. Essa è considerata una genetica, anomala reattività a un farmaco e propone continuità di studi rivolti a chiarirne sempre meglio la natura.
In conformità a quanto riportato, è bene ricordare che nelle reazioni di tipo I gli allergeni si combinano con le IgE specifiche, legate ai recettori di membrana sulle mast-cellule tissutali e sui basofili ematici, conseguendone una reazione antigene-anticorpo. Si verifica, quindi, un rapido rilascio di potenti mediatori vasoattivi e infiammatori in parte preformati, come l’istamina e la triptasi, oppure di nuova sintesi dai lipidi di membrana, come i leucotrieni e le prostaglandine. Anche le citochine pro infiammatorie, come l’interleuchina 4 e l’interleuchina 13 vengono rilasciate in poche ore dalle mast-cellule e dai basofili, causando vasodilatazione, aumento della permeabilità capillare, ipersecrezione ghiandolare, contrazione della muscolatura liscia e infiltrazione nei tessuti di eosinofili e di altre cellule infiammatorie. Nelle reazioni di tipo II citotossiche, invece, l’anticorpo reagisce con le componenti antigeniche della cellula o dei tessuti, oppure con un antigene o un aptene, legati a una cellula o a un tessuto. La reazione antigene-anticorpo può attivare le cellule killer T o i macrofagi, determinando la citotossicità cellulo-mediata anticorpo-dipendente e inclusiva, quasi sempre, dell'attivazione del complemento che può provocare l'adesione opsoninica, mediante il rivestimento della cellula con l'anticorpo. Ne consegue l’attivazione dei componenti del complemento per mezzo del C3, con fagocitosi della cellula o con l'attivazione di tutto il sistema complementare, risultandone la citolisi o il danno tissutale.
Nelle reazioni di tipo III gli IC (immunocomplessi) solubili, circolanti si depositano nei vasi o nei tessuti, attivando il complemento e innescando, così, una reazione infiammatoria acuta che porta alla migrazione dei polimorfonucleati e al rilascio di enzimi proteolitici lisosomiali e dei fattori di permeabilità nei tessuti. L’eccesso di anticorpo fa precipitare rapidamente gli IC, dove è localizzato l'antigene, oppure si ha la loro fagocitosi da parte dei macrofagi con rimozione del danno. In presenza, invece, di un lieve eccesso di antigene, gli IC tendono a essere più solubili, con capacità di determinare, per la loro deposizione nei tessuti, reazioni sistemiche.
Infine, le reazioni di tipo IV d’ipersensibilità cellulare, cellulo-mediate, ritardate o di tipo tubercolinico, sono prodotte dai linfociti T, sensibilizzati dal contatto con un antigene specifico. In questo caso, però, il danno tissutale non si produce necessariamente. Per la presenza degli anticorpi circolanti, questa forma di reazione può essere trasmessa dagli individui sensibilizzati ai non, tramite i linfociti del sangue periferico, ma non attraverso il siero. I linfociti T, sensibilizzati, innescati o attivati dal contatto con un antigene specifico, possono indurre il danno immunologico con un effetto tossico diretto o attraverso la liberazione di sostanze solubili, le linfochine. I T linfociti, attivati nelle colture tissutali, dopo la sensibilizzazione, distruggono le cellule bersaglio per contatto diretto e, per la liberazione di diverse citochine, influenzano l'attività dei macrofagi, dei neutrofili e delle cellule killer linfoidi.
Una buona conoscenza delle proprietà dei farmaci è la base per evitare quanto più possibile le ADR (Adverse Drug Reactions), che rappresentano un rilevante problema di salute pubblica. Secondo diverse fonti, circa il 5-10% dei pazienti in trattamento farmacologico è colpito da una reazione avversa. Questo evento, peraltro, causa circa il 5% dei ricoveri ospedalieri della popolazione totale e oltre il 15%nei pazienti anziani. La loro incidenza, poi, in ospedale è stimata superiore al 10%, con quote non basse di casi particolarmente gravi, tanto da poter portare al decesso. Una recente stima negli USA ha classificato le ADR tra la quarta – sesta causa di morte, con un impatto rilevante sui costi, corrispondenti a 30 - 130 miliardi di dollari in un anno, superiori a quelli determinati dal diabete.
L’errore in medicina nell’uso dei farmaci risulta, di poi, spesso connesso alle ADR che rappresentano l’evento avverso sempre più frequente nella sanità. In una recente analisi ospedaliera la prescrizione, in particolare, si presentava come la critica fase responsabile di circa il 60% di errore, con un consequenziale risultato di aumento delle degenze e dei costi aggiuntivi sino al 2%. Eppure, la reazione avversa, spesso definita inevitabile, potrebbe essere prevenuta con un monitoraggio più puntuale e aggiornato della farmacocinetica e farmacodinamica dei farmaci, riconoscendo tempestivamente le condizioni predisponenti, valutando il pericolo riguardante il meccanismo d’azione delle singole sostanze e delle loro potenziali interazioni. L'Organizzazione Mondiale della Sanità ha fornito la seguenteclassificazione eziologica delle ADR:
- Tipo A: dose-correlata; farmacologicamente prevedibile.
-
Tipo B: Non-dose-correlata, bizzarra e imprevedibile,
- a. immunocorrelata, come le reazioni di ipersensibilità,
- b. non immunocorrelata, come la porfiria, l’ipertermia maligna, la sindrome neurolettica maligna (poiché i meccanismi di queste reazioni specifiche sono ora meglio comprese, esse possono essere riclassificate come di tipo A).
- Tipo C: dose-tempocorrelata, legata alla durata e al dosaggio di esposizione, come la soppressione ipotalamo-ipofisi-surrene della terapia con glucocorticoidi.
- Tipo D: tempocorrelata, ritardata, come nella discinesia tardiva.
- Tipo E: da sospensione, come in caso di stupefacenti o beta-bloccanti.
- Tipo F: da errore imprevistodella terapia in rapporto a possibile interazioni farmacologiche, come nel fallimento dei contraccettivi orali a causa dell'induzione di enzimi con un secondo farmaco.
Da notare che i tipi A e B sono stati proposti negli anni ’70, mentre gli altri successivamente, per integrazione necessaria dei primi due.
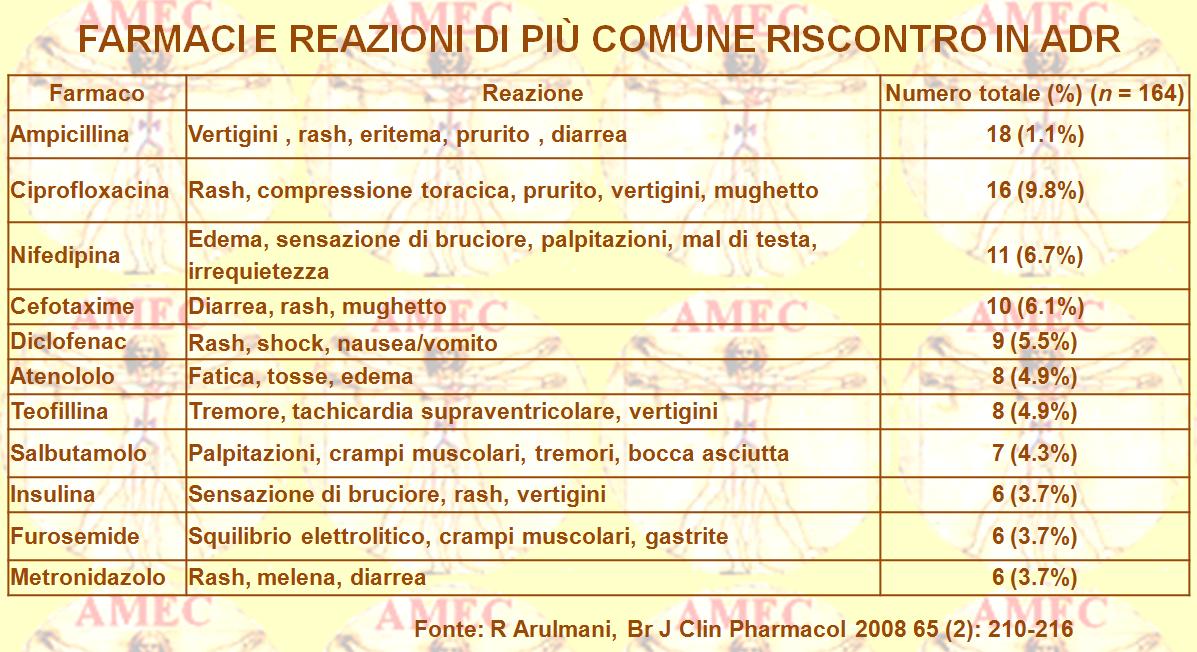
Di un indubbio interesse è il dato che alcune interazioni sono dovute al sistema di enzimi del citocromo P-450, utilizzati dall’organismo per rimuovere un farmaco secondo interazioni, anche complesse, con aumento o riduzione della sua attività. È il caso della teofillina e della ciprofloxacina che determinano un’up-regulation della via del citocromo, che cancella anche gli estrogeni e la fenitoina. In tal modo, i contraccettivi orali e gli anticonvulsivanti possono fallire la loro azione per una loro rapida eliminazione, senza che si possa realizzare un loro livello adeguato nel sangue. D’altro canto, gli inibitori delle monoamino ossidasi possono provocare ipertensione fatale nei pazienti che hanno consumato cibi contenenti alte concentrazioni di tiramina, come il vino Chianti, alcuni pesci affumicati e formaggi stagionati.


.png)





